1. Welcome to GSEAPY’s documentation!
1.1. GSEAPY: Gene Set Enrichment Analysis in Python.

1.2. GSEApy is a Python/Rust implementation of GSEA and wrapper for Enrichr.
It’s used for convenient GO enrichments and produce publication-quality figures from python.
GSEApy could be used for RNA-seq, ChIP-seq, Microarry data.
Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g. phenotypes).
The full GSEA is far too extensive to describe here; see
GSEA documentation for more information.
Enrichr is open source and freely available online at: http://amp.pharm.mssm.edu/Enrichr .
1.3. Citation
Zhuoqing Fang, Xinyuan Liu, Gary Peltz, GSEApy: a comprehensive package for performing gene set enrichment analysis in Python,
Bioinformatics, 2022;, btac757, https://doi.org/10.1093/bioinformatics/btac757
1.4. Installation
Install gseapy package from bioconda or pypi.
# if you have conda
$ conda install -c bioconda gseapy
# or use pip
$ pip install gseapy
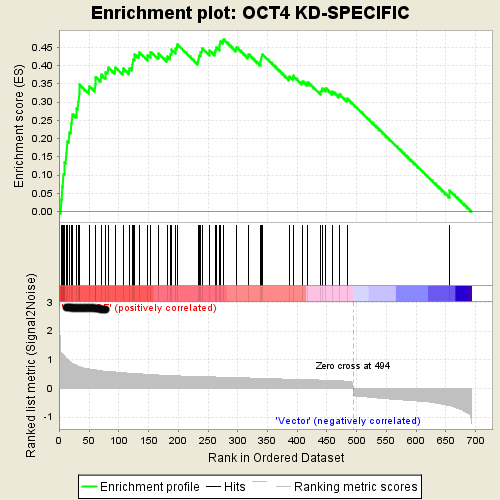
1.5. GSEA Java version output:
This is an example of GSEA desktop application output
1.6. GSEApy Prerank module output
Using the same data from GSEA, GSEApy reproduces the example above.
Using Prerank or replot module will reproduce the same figure for GSEA Java desktop outputs
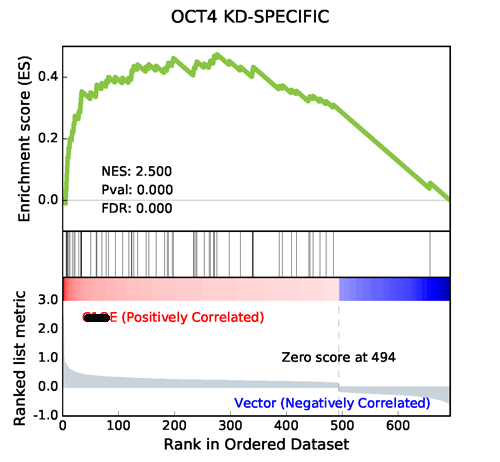
Generated by GSEAPY
GSEApy figures are supported by all matplotlib figure formats.
You can modify GSEA plots easily in .pdf files. Please Enjoy.
1.7. GSEApy enrichr module
A graphical introduction of Enrichr
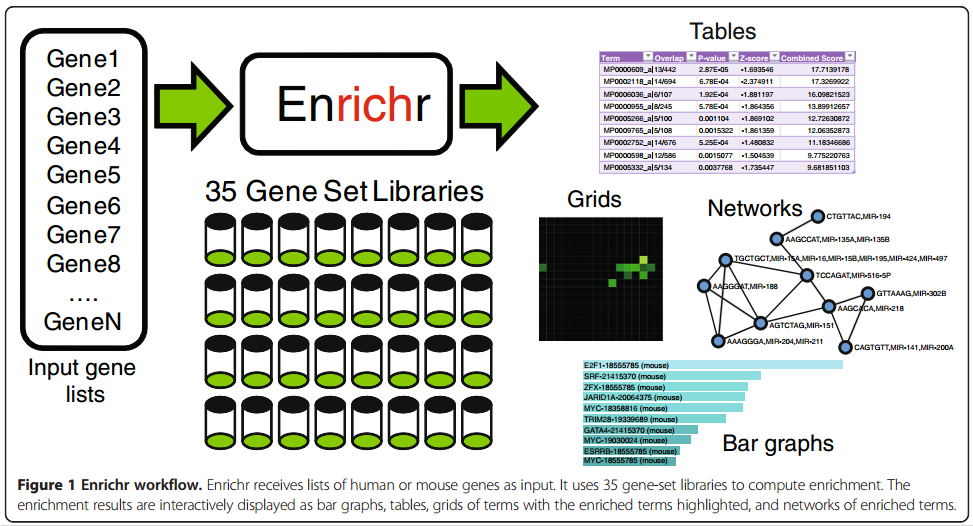
The only thing you need to prepare is a gene list file in txt format(one gene id per row), or a python list object.
Note: Enrichr uses a list of Entrez gene symbols as input. You should convert all gene names to uppercase.
For example, both a list object and txt file are supported for enrichr API
# if you prefer to run gseapy.enrchr() inside python console, you could assign a list object to
# gseapy like this.
gene_list = ['SCARA3', 'LOC100044683', 'CMBL', 'CLIC6', 'IL13RA1', 'TACSTD2', 'DKKL1',
'CSF1', 'CITED1', 'SYNPO2L']
# an alternative way is that you could provide a gene list txt file which looks like this:
with open('data/gene_list.txt') as genes:
print(genes.read())
CTLA2B
SCARA3
LOC100044683
CMBL
CLIC6
IL13RA1
TACSTD2
DKKL1
CSF1
CITED1
SYNPO2L
TINAGL1
PTX3
1.8. Installation
# if you have conda
$ conda install -c conda-forge -c bioconda gseapy
# or use pip to install the latest release
$ pip install gseapy
For API information to use this library, see the Developmental Guide.